YALEPIC® 通用型无缝克隆重组试剂盒
基于同源重组原理的一步法通用型等温无缝克隆试剂盒,属于非连接酶依赖型体系。可将含15~25bp同源臂的1~11个DNA片段在50℃、5~15分钟内定向重组至任意载体的任意位点,不受传统酶切位点限制,载体自连背景极低。含阳性对照,适合高通量克隆与自动化平台应用。
| 产品货号 | 产品规格 | 目录价 | 促销价 | 购物车 |
|---|---|---|---|---|
| YTD38001 | 100rxns | 1000.00 | — | + |
- 产品详情
- FAQ
- COA
- 相关文献
产品简介
YALEPIC® Universal Seamless Cloning Assemble Kit是基于同源重组原理开发的一步法单片段/多片段通用型等温无缝克隆试剂,属于非连接酶依赖型体系,载体自连背景极低,可显著提高克隆的重组效率及对杂质的耐受程度,制备的高纯度线性化载体和插入片段可不纯化直接用于重组克隆。本产品可快速、定向、准确的将含有载体末端重叠区域的插入片段(单个片段,或多至11个顺序拼接的片段)定向重组至任何载体的任何位点,50℃,5 ~ 15 min 即可完成重组,不受酶切位点限制,载体末端与插入片段末端以及相邻插入片段末端之间需要 15~25 bp 能够相互同源重组的完全一致的序列。本产品具有高保真、高效、便捷的DNA连接性能适用于文库构建等应用,阳性克隆率高达 95%。
产品优势
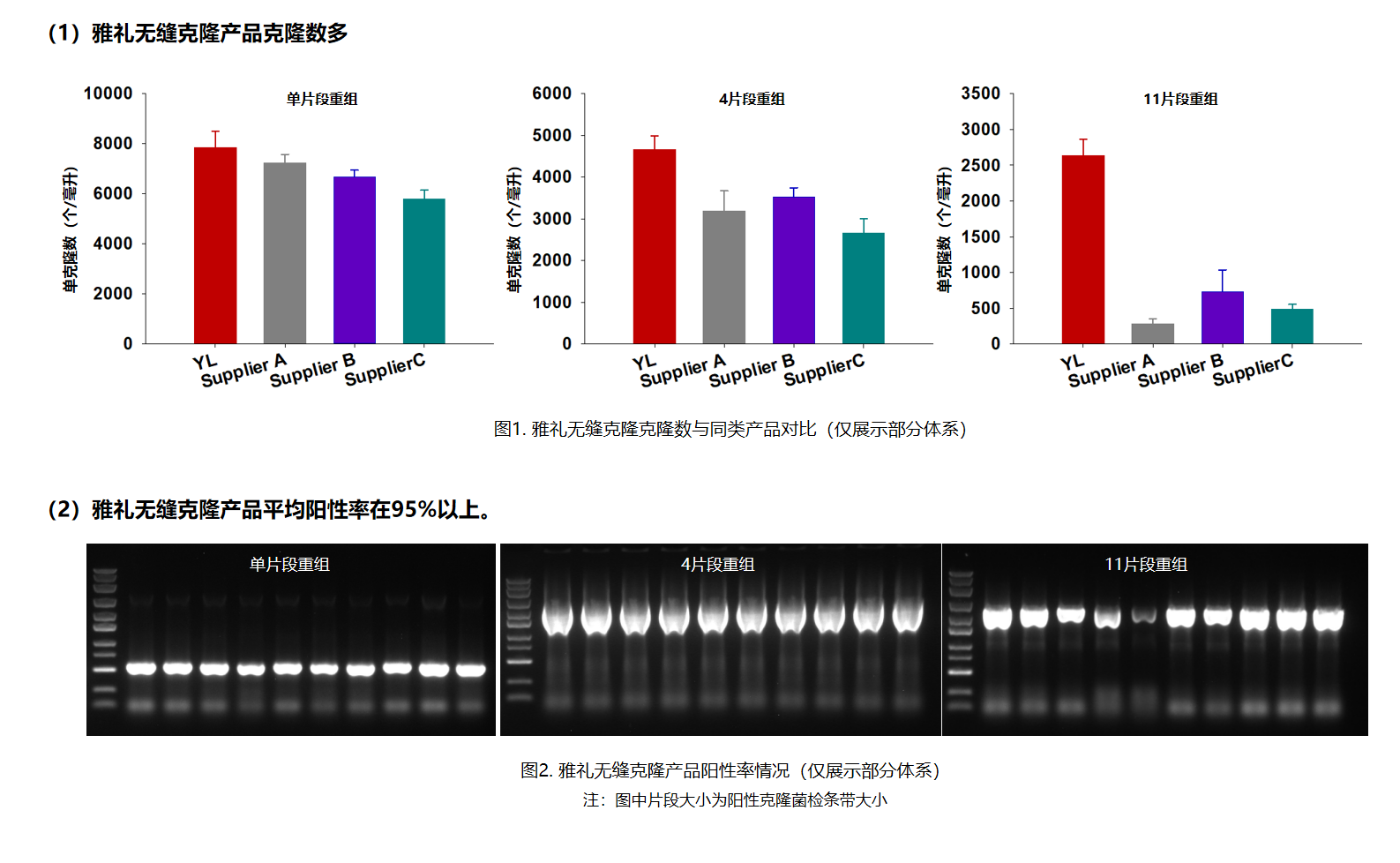
- 高保真性,克隆数适中、克隆阳性率>95%;
- 高效组装,50℃,5~15min完成重组;
- 兼容性高,可组装1~11个DNA片段;
- 操作简便,Mix和样品等体积混匀即可。
适用范围
- 单片段/多片段定向无缝克隆(1–11个片段)
- 高通量克隆与自动化平台应用
- 重组蛋白表达载体构建
- 多位点定点突变
- 基因文库构建
- CRISPR相关载体构建
实验案例
保存及运输条件
– 20℃ 储存,≤ 0℃ 运输。
FAQ
Q1:平板上很少或没有克隆菌落,该怎么办?
A:可能原因及解决方案:
- 引物设计错误——同源臂长度应为15–25 bp,GC含量控制在40%–60%之间。若GC含量高于70%或低于30%,重组效率会受到较大影响。建议重新设计引物。
- 感受态细胞效率低——使用新鲜制备或妥善冻存的感受态细胞,确保转化效率>10⁸ cfu/μg。连接产物体积不应超过感受态细胞体积的1/10,否则会降低转化效率。
- DNA投入量不足或比例不佳——载体与插入片段的最佳摩尔比为1:2。推荐使用公式快速计算:载体用量(ng)≈ 0.02 × 载体碱基数;片段用量(ng)≈ 0.04 × 片段碱基数。
- DNA纯度不够——若使用未纯化的DNA,其总体积不应超过反应体系体积的1/5(如20 μL体系不超过4 μL)。建议进行凝胶回收纯化,纯化产物溶解于ddH₂O中。
- 试剂盒本身问题——建议先使用试剂盒附带的阳性对照进行实验,排除试剂盒质量问题-。若阳性对照有克隆,则说明试剂盒正常。
Q2:克隆菌落很多,但测序发现插入片段有碱基突变或缺失,是什么原因?
A:出现这种情况通常不是试剂盒本身的问题,而是由于PCR扩增引入了突变。可能原因及解决方案如下:
- PCR过程引入了突变——建议使用高保真DNA聚合酶进行目的片段的扩增,避免使用普通Taq酶。
- 非特异性PCR产物混入——通过凝胶电泳检查PCR产物的条带特异性。如果存在多个PCR产物,会导致载体中连入错误的插入片段,建议对PCR产物进行凝胶纯化后再用于重组反应。
- 载体线性化不彻底——即使是痕量未完全酶切的环状载体残留,也会产生很高的转化背景。可通过阴性对照检测载体是否线性化完全,优化酶切体系(提高限制性内切酶用量、延长酶切时间或胶回收酶切产物)。
Q3:同源臂应该设计多长?有什么设计原则?
A:设计原则:
- 同源臂长度:18–25 bp最为理想。若少于15 bp,重组效率会明显下降。比推荐长度少几个碱基(如14 bp)仍可能工作,但效率会受到影响。
- GC含量:同源臂区域的GC含量应控制在40%–60%之间。若GC含量高于70%或低于30%,重组效率会受到较大影响。
- 序列特异性:应避免选择克隆位点上下游50 bp内含有重复序列的区域,否则可能导致非特异性重组。
- 引物设计通式:5′-[同源序列(15–25 bp)]-[酶切位点(可选)]-[基因特异性扩增引物]-3’。
- 注:根据产品说明书,本试剂盒要求载体末端与插入片段末端以及相邻插入片段末端之间需要15–25 bp能够相互同源重组的完全一致序列。
Q4:同源重组和无缝克隆到底是什么关系?有什么区别?
A:一句话结论:同源重组是方法,无缝克隆是结果。同源重组是新一代定向克隆技术,不依赖酶切和连接步骤,依靠DNA片段与线性化载体末端15–20 nt同源序列完成重组,可将片段克隆至任意位点,载体自连背景极低。
Q5:为什么会出现大量假阳性克隆(克隆不含插入片段)?如何避免?
A:可能原因及解决方案:
- 载体线性化不完全——即使是痕量未完全酶切的环状载体也会产生很高的转化背景。建议:①提高限制性内切酶用量;②延长酶切反应时间;③进行胶回收纯化酶切产物;④设置阴性对照检测载体是否线性化完全。
- 相同抗性的环状质粒污染——当以环状质粒为模板进行插入片段PCR扩增时,残留的环状质粒模板会产生较高的转化背景。建议:①使用预线性化质粒作为扩增模板;②对PCR产物进行DpnI消化(DpnI可识别甲基化位点,切割模板质粒);③对扩增产物进行胶回收纯化。
- 平板抗性不足——确保使用正确的抗生素以及新鲜制备的抗生素平板。
Q6:50°C反应时间需要多久?不同片段数量如何调整?
A:推荐反应时间:1–2个片段:5–15分钟;3–5个片段:15–30分钟;载体>10 kb或片段>4 kb:延长至30–60分钟。注意严格控制反应温度(50°C),温度偏差会影响重组酶的活性,若反应液不慎沾到管壁上,可通过短暂低速离心使其沉入管底。
Q7:PCR产物是否需要纯化?未纯化PCR产物能否直接使用?
A:可以,但需注意以下限制:
- 若线性化载体与插入片段已经过纯化,且经电泳检测条带单一或无Smear残留时,可直接使用超微量核酸蛋白检测仪进行浓度测定(A260/A280在1.8–2.0之间时浓度值可信)。
- 若为未纯化的DNA,其使用总体积不应超过反应体系体积的1/5(如20 μL体系不超过4 μL)。但需要注意:EDTA等金属离子螯合剂会抑制无缝克隆反应,因此纯化产物应溶解于ddH₂O中,切勿使用Tris-EDTA等缓冲液进行溶解。
- 建议: 为保证最佳重组效率,推荐对线性化载体和插入片段扩增产物进行凝胶回收纯化。
Q8:电转感受态细胞能否用于无缝克隆反应?
A:不建议使用电转化感受态细胞。推荐做法: 使用高质量的化学感受态细胞(转化效率>10⁸ cfu/μg),重组产物加入感受态细胞不超过1/10体积,以避免转化抑制。
Q9:如何验证重组反应是否成功?
A:推荐以下验证方法:
- 阳性对照——建议使用试剂盒附带的阳性对照进行平行实验。若阳性对照有克隆并检测为阳性,则可排除试剂盒本身的问题。
- 菌落PCR验证——使用载体的通用引物或至少一条通用引物进行菌落PCR检测。
- 提取质粒进行双酶切验证——从阳性克隆中提取质粒,使用预期位点的限制性内切酶进行双酶切鉴定。
- 测序验证——最终的阳性克隆建议进行测序验证,确认插入片段序列正确无误。
Q10:本试剂盒最多可以组装多少个片段?总片段大小有限制吗?
A:本产品可高效组装1–11个DNA片段。多片段组装时,建议先尝试较少片段(2–3个)的构建以优化实验条件,再逐步增加片段数量。
Q11:试剂盒应该如何保存和运输?反复冻融有影响吗?
A:如下:
- 保存条件: -20°C储存。运输条件: ≤0°C运输。
- 建议避免或减少反复冻融。每次使用前应充分溶解并轻轻混匀,使用后立即放回-20°C保存,取用时应使用无核酸酶的枪头和离心管。本产品可反复冻融30次不显著降低性能。
- 使用前如发现试剂略有冻结粘稠,冰上解冻后应为澄清透亮液体,不影响正常使用。
Q12:菌落PCR检测无目的条带,但克隆有空质粒条带,是什么原因?
A:可能原因: 重组不成功,载体线性化不完全。
解决方案:
- 重新检查线性化载体的酶切是否彻底,确保载体完全线性化
- 优化酶切体系:提高限制性内切酶的用量,延长酶切反应时间
- 对酶切产物进行胶回收纯化,去除残留的环状质粒
- 建议使用经过验证的载体通用引物进行菌落PCR验证
